Why Targeting Prostaglandin Signalling Reawakens the Entire Antitumor Immune Response
Clinical validation of the COX2–PGE2/PGD2 axis is moving from epidemiology to mechanism — and reframing how we think about immune-suppressive tumors.
Immune checkpoint inhibitors transformed oncology, but the ceiling is real. Across most solid tumors, fewer than 40% of patients respond durably to anti-PD(L)1 therapy. The reason isn't mysterious: blocking PD-1 only releases the brake on T cells. If the tumor microenvironment is simultaneously suppressing every other arm of the immune system — NK cells, dendritic cells, the myeloid compartment, regulatory T cells — releasing one brake is not enough.
This is where the prostaglandin axis has re-emerged as one of the most clinically grounded targets in immuno-oncology. Decades of epidemiological data on aspirin and NSAIDs already suggested that interfering with prostaglandin production reduces cancer incidence and mortality. What's new is mechanistic clarity: we now understand how prostaglandins shape the immunosuppressive landscape across distinct tumor compartments, and why broad blockade of this axis — rather than narrow inhibition of a single receptor — may finally translate into clinical benefit. It is also what drove us to develop OKN4395, a first-in-class triple EP2/EP4/DP1 antagonist designed to silence the entire axis at once.
One pathway, every immune compartment
The cyclooxygenase-2 (COX2) enzyme generates two key lipid mediators implicated in cancer immunosuppression: prostaglandin E2 (PGE2) and prostaglandin D2 (PGD2). Both signal through Gs-coupled receptors — EP2 and EP4 for PGE2, DP1 for PGD2 — and both converge on the same intracellular second messenger: cyclic AMP (cAMP). Elevated cAMP in immune effector cells is one of the most established mechanisms of immune suppression in biology.
What makes this axis particularly damaging in tumors is its reach. PGE2 and PGD2 together don't suppress only one immune compartment, they suppress all of them simultaneously. And the suppression is precise rather than blunt: across every compartment studied, the effector machinery itself is left intact — T cells still recognize their targets, NK cells still load their cytotoxic granules, dendritic cells still capture antigen — while one specific stage of effector deployment is selectively disabled.
T cells. PGE2 directly inhibits CD4+ and CD8+ T cell proliferation, IFNγ production, and cytotoxic function. Work published in Nature in 2024 identified the mechanism: PGE2 disrupts IL-2 receptor signaling and impairs mitochondrial function in tumor-infiltrating lymphocytes, limiting expansion of the stem-like CD8+ T cell populations that drive durable anti-tumor immunity. TCR signaling itself is preserved; what fails is the metabolic and cytokine support needed for clonal expansion. In functional assays, PGE2 fully abolishes IFNγ secretion from CD8+ T cells, and only dual EP2/EP4 blockade fully restores it, whereas single receptor inhibition is compensated by signaling through the other. Critically, this suppression is independent of the PD-1 axis: in our recent preprint, PGE2 fully suppresses IFNγ production in dendritic-cell-T-cell co-cultures whether or not anti-PD-1 is present (checkpoint blockade cannot rescue the function PGE2 removes) while triple EP2/EP4/DP1 antagonism with OKN4395 restores it additively with anti-PD-1. PGD2 signaling through DP1, which human T cells express at substantial levels, raises intracellular cAMP to the same magnitude as PGE2 and suppresses CD8+ T cell IFNγ with comparable potency, and triple EP2/EP4/DP1 antagonism with OKN4395 reverses this suppression as effectively as dedicated DP1 inhibitors. Because both prostaglandins converge on the same second messenger, blocking PGE2 alone leaves a fully functional parallel suppressive channel open.
NK cells. PGE2 acts at multiple stages of the NK antitumor response. It selectively downregulates the activating receptors NKG2D, NKp30, and NKp44, while leaving inhibitory receptors unaffected, and disrupts downstream activating signaling independently of receptor expression, reducing cytotoxic output. The recognition machinery is intact, but the signal to kill is not. PGE2 also redirects NK cell trafficking by downregulating CXCR3 and upregulating CXCR4, rerouting NK cells away from tumors and toward the bone marrow. PGD2 produces a parallel suppressive effect on NK cytotoxicity through DP1, and NK dysfunction propagates downstream, since NK cells recruit type 1 conventional dendritic cells (cDC1) into the tumor.
Dendritic cells. Tumor-derived PGE2 signaling through EP2 and EP4 programs intratumoral cDC1s into a dysfunctional state via cAMP-driven loss of the master transcription factor IRF8. The consequences are strikingly selective: PGE2-exposed cDC1s retain the ability to acquire tumor antigens, traffic to draining lymph nodes, and prime naïve CD8+ T cells normally. What they lose is the capacity to produce CXCL9 and IL-12 within the tumor itself, the two signals required to recruit primed CD8+ T cells into the tumor bed and expand them locally into effectors. Genetic ablation of EP2 and EP4 specifically in cDC1s restores this function and achieves CD8+ T cell-dependent tumor rejection in otherwise progressing models. In parallel, PGE2 reprograms mature regulatory dendritic cells (mregDCs) to produce CCL17 and CCL22, actively recruiting regulatory T cells and stabilising their suppressive Foxp3+ phenotype. The dendritic cell compartment thus serves as a dual amplifier: cDC1 dysfunction silences the local CD8+ T cell response while mregDC reprogramming imports and stabilizes the suppressor compartment in the same tissue.
Myeloid compartment. Across tumor-associated neutrophils, MDSCs and macrophages, PGE2-EP2/EP4 signaling drives pro-inflammatory and angiogenic genes (IL1B, PTGS2, VEGFA, HIF1A, CXCL2) creating the paradox of the tumor microenvironment: active inflammation that sustains tumor growth rather than immune activation. The mechanism has been resolved in monocytic MDSCs: tumor-derived PGE2 acting through EP2 induces nuclear p50 NF-κB, transcriptionally redirecting IFNγ-driven STAT1 recruitment toward NOS2 and away from TNFα, so that IFNγ stimulation produces nitric oxide-mediated T cell suppression rather than inflammatory output. This has been validated clinically: circulating M-MDSCs from colorectal cancer patients show elevated nuclear p50, NOS2 and EP2, and EP2 antagonism reprograms M-MDSCs toward a NOS2-low/TNFα-high phenotype with antitumor efficacy comparable to anti-PD-1 in vivo. The same regulatory node drives distinct, indication-specific programs: in pancreatic ductal adenocarcinoma, PGE2-TNF synergy elicits an IL-1β+ macrophage state from the earliest stages of tumorigenesis (Caronni et al., Nature 2023); and in melanoma, a protumoral macrophage subset uses HPGDS to generate PGD2 that excludes CD8+ T cells from the tumor core through paracrine DP1 signaling — these PGD2-high macrophages are enriched in anti-PD-1 non-responders.
The picture that emerges is unusual in immuno-oncology: a single regulatory node that simultaneously dampens innate effector function, silences local antigen presentation, recruits and stabilizes suppressor cells, and converts cytokine-driven immune programs into suppressive ones, all while leaving the effector machinery intact at each step.
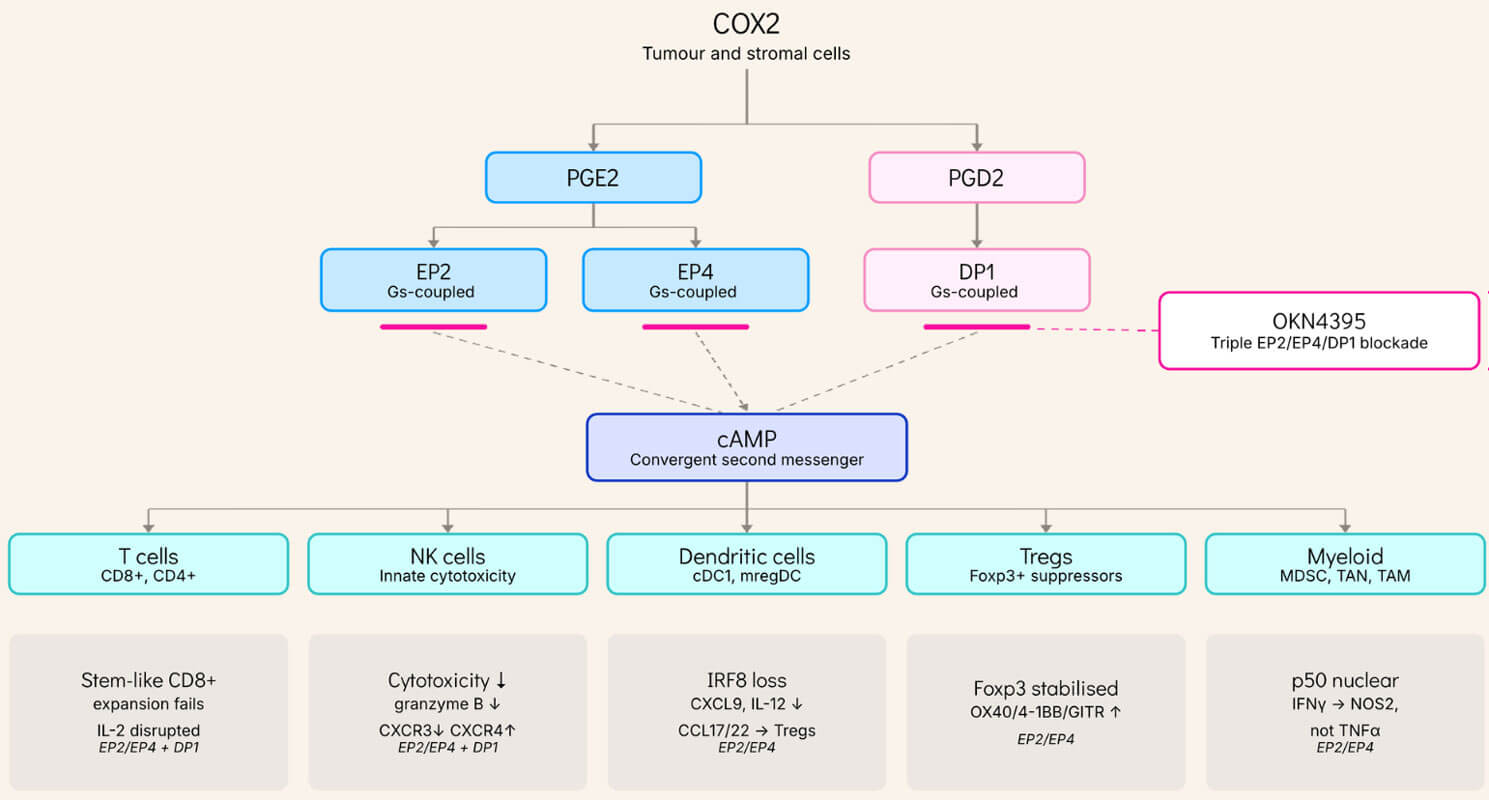
OKN395 blocks all three Gs-coupled receptors, preventing cAMP elevation across compartments.
Box tag = receptor(s) with evidence in that compartment.
PGE2/EP2-EP4 dominates dendritic, Treg and myeloid; PGD2/DP1 adds to T-cell and NK.
Why redundancy demands broad blockade
The mechanistic redundancy of this axis explains both why single-receptor blockade produces real but bounded benefit and why broader blockade should capture more. The most direct clinical test to date is ONO-4578-08, a randomized, double-blind phase 2 trial in which the selective EP4 antagonist ONO-4578, added to nivolumab and chemotherapy in first-line HER2-negative gastric/gastro-oesophageal junction cancer, significantly prolonged progression-free survival (HR 0.67) with favorable OS over nivolumab and chemotherapy alone (Nakayama et al., JCO 2026). This is the first randomized evidence that blocking the prostaglandin brake on top of checkpoint inhibition improves outcomes in a solid tumor, a direct clinical validation of the premise on which this entire axis rests. Yet the benefit was concentrated in PD-L1 CPS≥1 tumors and was achieved by antagonising a single receptor: EP4 blockade is compensated by PGE2 signaling through EP2 in preclinical models, and the parallel PGD2–DP1 axis is left entirely untouched. Dual EP2/EP4 blockade is required to fully abolish PGE2-driven immunosuppression, and even then, DP1 remains an open escape route.
PGE2 transcriptionally upregulates the adenosine receptors A2AR and A2BR on CD8+ T cells, priming them for a parallel wave of cAMP-driven suppression delivered by CD73/CD38-generated adenosine; combined COX2 and CD38 blockade reverses anti-PD-1 resistance in vivo where either alone fails. This is direct evidence that these brakes are non-redundant, and that relieving one leaves the others intact.
Upstream COX2 inhibition with NSAIDs is broad enough to block all of these pathways simultaneously, which is precisely why COX2 inhibitors have produced the strongest epidemiological and clinical signals to date. But COX2 inhibitors also block prostaglandin pathways essential for normal tissue homeostasis, which has constrained their use as chronic oncology agents.
The therapeutic question is whether one can capture the breadth of COX2 inhibition at the receptor level while avoiding systemic toxicity. In primary human immune cell assays, OKN4395 fully restores T cell and NK cell function under both PGE2 and PGD2 pressure, with no activity at structurally related prostanoid receptors (EP1, EP3, IP, FP, TP, DP2) or across a broader panel of 166 GPCRs. Triple blockade closes the redundancy loop.
From epidemiology to prospective validation in colorectal cancer
The strongest test of any therapeutic hypothesis is a prospective, biomarker-driven randomized trial. For the COX2–PGE2 axis, that test has been done in colorectal cancer — and the results have moved the pathway from the chemoprevention literature into the adjuvant treatment paradigm.
The mechanistic basis is well established. COX2-driven PGE2 activates the PI3K–AKT–mTOR axis in tumor cells, and PI3K signaling reciprocally upregulates COX2 expression, creating a self-amplifying loop. Approximately 40% of resected colorectal cancers carry activating PI3K pathway alterations, a molecular subset for which the pathway is oncogenically central, not merely contributory.
The ALASCCA trial, published in the New England Journal of Medicine in 2025, randomized 626 patients with stage I–III colorectal cancer harboring somatic PI3K pathway alterations to 160 mg aspirin or placebo for three years. The 3-year cumulative incidence of recurrence dropped from 14.1% to 7.7% in patients with PIK3CA hotspot mutations (HR 0.49), and from 16.8% to 7.7% in patients with other PI3K pathway alterations (HR 0.42).
A second dataset extends the picture beyond genomic selection. A post-hoc analysis of CALGB/SWOG 80702, published in JAMA Oncology in 2026, used tumor-informed ctDNA to stratify 940 patients with resected stage III colon cancer by post-operative molecular residual disease. Among ctDNA-positive patients, adjuvant celecoxib reduced both recurrence and death versus placebo (DFS HR 0.61; OS HR 0.62), translating to an 18-point improvement in 3-year disease-free survival. Among ctDNA-negative patients, celecoxib added nothing. Critically, the effect was preserved in MSI-high and PIK3CA-wildtype tumors, indicating that MRD-positivity captures a dimension of pathway dependence that is at least partially independent of PIK3CA genotype: most plausibly the inflammatory and myeloid composition of the residual microenvironment from which recurrence emerges.
Read together, these trials make a coherent claim. COX2 inhibition is therapeutically actionable in colorectal cancer, provided the right patients are selected. Two non-overlapping biomarkers — somatic PI3K pathway alteration and post-operative ctDNA positivity — independently identify subsets that benefit. The question is no longer whether to drug this pathway, but in whom.
Aspirin and celecoxib confirm that interrupting COX2 prostaglandin signaling changes outcomes, but they do so by inhibiting prostaglandin synthesis upstream and indiscriminately, which is also why their use as chronic oncology agents is constrained by cardiovascular and gastrointestinal liabilities of blocking homeostatic prostanoids. Receptor-selective triple blockade is built to capture the immunosuppressive breadth of the axis — EP2, EP4 and DP1 — while leaving the homeostatic prostanoid receptors untouched, making sustained blockade feasible and extending the same mechanistic logic to indications beyond colorectal cancer.
The next inflection point: precision, not breadth
The COX2-PGE2/PGD2 axis is now one of the most clinically validated immuno-oncology pathways outside of the checkpoint axes themselves. Three decades of epidemiology, a coherent multi-compartment mechanism, and three prospective trials, including the first receptor-antagonist-plus-checkpoint trial, all converge. What the field has lacked until recently is a molecule with the breadth to silence the entire cAMP-driven axis without the off-target liabilities that retired the first generation of COX2 inhibitors, and a clinical strategy capable of matching that breadth to the patients most likely to benefit.
Both are now within reach. Triple EP2/EP4/DP1 antagonism reconstitutes the breadth of COX2 inhibition at the receptor level, sparing the prostanoid pathways implicated in cardiovascular homeostasis. And the colorectal cancer experience has established the operational template for patient selection: a genomic biomarker identifies tumors hardwired into pathway dependence, while a microenvironmental biomarker identifies tumors whose residual immune landscape is pathway-driven. The same logic generalizes to the next-conviction indications, those where checkpoint inhibition has produced partial signals and where pathway-driven dysfunction of cDC1s, neutrophils, macrophages, MDSCs, or Treg dominance has been shown to predict resistance to anti-PD-1, and where myeloid and Treg compartments dominate the microenvironment. In each of these settings, the equivalent of PIK3CA genotyping or post-operative ctDNA either already exists or can be constructed from the same mechanistic biology. Increasingly, that construction relies on K Pro’s multimodal AI analyses that integrate genomic, transcriptomic, and pathology-derived features into a coherent pathway activity score.
The inflection point ahead is not whether prostaglandin blockade will enter the clinic, but how precisely it is deployed when it does. The compartments tell us which immune defects to expect. The biomarkers tell us in which patients those defects are doing the work. OKN4395 tells us the breadth required to neutralize them. Bringing all three to bear at once is now an executable strategy.
Authors


Testimonial
